Abstract
The world is in an immediate need of treatment for coronavirus disease (COVID‐19). Chronic exposure of hydroxychloroquine in the treatment of COVID‐19 may have multiple adverse effects on human physiology, such as cardiac arrhythmias. Natural compounds need to be evaluated as treatment and preventive agents in coronavirus infection. A total of 30 compounds of Solanum tuberosum and Brassica juncea residue smoke water were selected for the virtual screening against SARS‐CoV‐1, SARS‐CoV‐2 and cellular proteins involved in the mechanism of infection. Docking analysis identified lead molecules with favorable binding energy, number of poses and hydrogen bond interactions, which indicates the effective modulation of ACE2 and TMPRSS2 receptors. Results indicated (a) curcumenol, (b) N ‐desmethylselegiline, (c) phentermine and (d) sphingolipid derivatives as a selective and potent candidates in comparison to hydroxychloroquine for COVID‐19 treatment. Our in silico findings, therefore, warrant further in vitro validations of the selected compounds for the discovery of novel preventive and therapeutic drug against SARS‐CoV‐2 infection.
1 INTRODUCTION
The world is going through heavy healthcare and socio‐economic burden of severe acute respiratory syndrome coronavirus2 (SARS‐CoV‐2) pandemic. Therapeutic/preventive target to control the COVID‐19 prevalence is the biggest challenge, globally. Initially, the new virus was named 2019‐nCoV, but subsequently the International Committee on Taxonomy of Viruses (ICTV) termed it as SARS‐CoV‐2 due to similarity with the causing agent of SARS outbreak (SARS‐CoVs) (Gorbalenya et al., 2020). SARS‐CoV‐2 was reported by The Chinese Center for Disease Control (China CDC) reported a cluster of severe pneumonia cases of unknown seafood and wet animal wholesale market in Wuhan, China (Li et al., 2020; Mackenzie & Smith, 2020). The outbreak was prominent and highly contagious, which remained the reason to evoke targeted and efficient therapy to control COVID‐19. The world is under an instantaneous need of finding proven and registered antiviral drugs or vaccines for COVID‐19 infection. Hygiene measures, as well as quarantine at home and social distance, reduce the rate of transmission, and post‐infection remedies are to be developed to prevent mortality. The treatment given to infected patients based on patient’s symptoms and broad‐spectrum antiviral drugs is the only option till new vaccine or therapeutic agents would be available throughout the world.
A specific set of proteins from human and SARS‐CoV are involved in the infection mechanism. Human transmembrane serine proteases type II (TMPRSS2) and angiotensin‐converting enzyme 2 (ACE2) play a main role in SARS‐CoV‐S protein binding in infection and entry into the human host cell (Jia et al., 2009; Shulla et al., 2011). Similarly, RNA‐dependent RNA polymerase (RdRp) presents a promising target due to its essential role in RNA synthesis and replication of RNA viruses (Shannon et al., 2020). Virus encoded proteases are actively involved in SARS‐CoV entry in host cells, and papain‐like protease (PLpro) is responsible in virus replication for the cleavages of replicase to release Nsp1, Nsp2 and Nsp3 (Harcourt et al., 2004). 3C‐like main protease (3CLpro) directly mediates the maturation of Nsps, which is essential in the life cycle of the virus (Wu et al., 2020). This can be the founding target to investigate the structural and catalytic mechanisms of SARS‐CoV‐2 3CLpro for drug development. Earlier, small peptide and molecules have been investigated as an inhibitor of SARS‐CoV and SARS‐CoV‐2 3CLpro (Wu et al., 2020). Two major proteins of COVID‐19, that is, RNA dependent RNA polymerase (RdRp) (Elfiky, 2020) and SARS‐CoV‐2 Spike protein (SARS‐CoV‐2‐S) (Bhattacharya et al., 2020) are the main proteins involved in attachment and infection cycle in human cell host. Whereas ACE2 and TMPRSS2 are involved in the mechanism of entry of COVID‐19 in host cell (Hoffmann et al., 2020). Molecular modulation by lead molecules can be utilized to target COVID‐19 through different proteins of virus and human to avert the infectivity of the virus. Through the tools of Bioinformatics and in silico methodologies, drug designing is accelerated to vanish the SARS‐CoV‐2 infection.
Currently, repurposing of hydroxychloroquine (HCQ) as a treatment of SARS‐CoV‐2 is in practice in India (COVID‐19, 2020) and in the rest of the world. Repurposing potential of HCQ against SARS‐CoV‐2 needs to be evaluated through in vivo and in vitro experiments thoroughly. In search of a superior drug (highly specific and minimum side effects) than HCQ, exploration of additional resources have to be performed.
Plants are a remarkable source of bioactive compounds, which require the in silico and wet lab experiments for its exploitation as a treatment for SARS‐CoV‐2. Smoke water of plant material is responsible for various physiological and biochemical activities in plant (Salomon et al., 2017). Solanum tuberosum and Brassica juncea are regular food crops all over the world and possess remarkable pharmacological potential (Bontempo et al., 2015; Lee et al., 2014; Rakholiya et al., 2011). In view of the above, the present research evaluates the in silico interaction study of reported molecules from Solanum tuberosum and Brassica juncea residue smoke water (Dave et al., 2018) docked with TMPRSS2, PLpro, SARS‐CoV‐2 3CLpro, SARS‐CoVRdRp, SARS‐CoV‐2‐S and ACE2 to prevent/cure COVID‐19.
2 MATERIALS AND METHODS
2.1 Ligand library
Phytoconstituents that possess remarkable pharmacological activity were selected as reported from Solanum tuberosum and Brassica juncea residue smoke water (Dave et al., 2018) and Ligand library was prepared of potential molecules to be used for the docking study with human, SARS‐CoV and SARS‐CoV‐2 proteins as given in Table 1.
| Sr. no. | Name | Structure | Application/property |
|---|---|---|---|
| 1aa Compound identified from Brassica juncea . | Curcumenol | 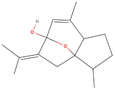 |
Anti‐inflammatory, Anti‐tumor and hepatoprotective (Lo et al., 2015) |
| 2aa Compound identified from Brassica juncea . | N‐Desmethylselegiline | 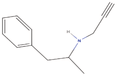 |
Treatment of Parkinson’s disease (Mizuta et al., 2000) |
| 3bb Compound identified from Solanumtuberosum . | Phentermine | 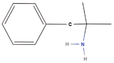 |
Appetite control (Baumann et al., 2000) |
| 4aa Compound identified from Brassica juncea . | Monodesmethylpheniramine |  |
Antihistamines are used in treatment of allergies (Kelmenson et al., 2013) |
| 5bb Compound identified from Solanumtuberosum . | N‐(3E‐hexadecenoyl)‐deoxysphing‐4‐enine‐1‐sulfonate | 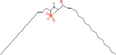 |
A sphingolipid (Cui et al., 2018) |
| 6bb Compound identified from Solanumtuberosum . | (5alpha,8beta,9beta)‐5,9‐Epoxy‐3,6‐megastigmadien‐8‐ol | 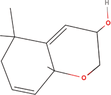 |
Constituent of Passifloraedulis (passion fruit) (Yannai, 2003) |
| 7bb Compound identified from Solanumtuberosum . | (R)‐2,3‐Dihydroxypropane‐1‐sulfonate | 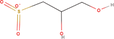 |
Metabolic intermediate of sulfoquinovose in plant (Mayer et al., 2010) |
| 8bb Compound identified from Solanumtuberosum . | 2‐(3‐Phenylpropyl) tetrahydrofuran | 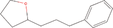 |
Flavouring compound (EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids, 2016) |
| 9bb Compound identified from Solanumtuberosum . | 3‐Nonyl‐1H‐pyrazole |  |
Found in plants, exhibits antimicrobial activity (Kumar et al., 2013) |
| 10bb Compound identified from Solanumtuberosum . | Mephentermine |  |
Treatment of hypotension (Weiner & Mason, 2019) |
| 11bb Compound identified from Solanumtuberosum . | N‐(2,4‐Eicosadienoyl) piperidine |  |
Alkaloid from Piper retrofractum (Yannai, 2003) |
| 12aa Compound identified from Brassica juncea . | Benzocaine |  |
Local anesthetic (Haas & Quinn, 2017) |
| 13aa Compound identified from Brassica juncea . | CAY10638 (5‐(4‐(2‐[Thiophen‐2‐yl]ethoxy)benzylidene)thiazolidine‐2,4‐dione) |  |
Anticancer (Li et al., 2009) |
| 14aa Compound identified from Brassica juncea . | Dimethylallyl diphosphate (DMAPP) | 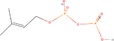 |
Important derivative of terpenoid biosynthesis (Wang et al., 2019) |
| 15aa Compound identified from Brassica juncea . | Î ± ‐9(10)‐EpODE | 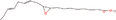 |
Plant and algae fatty acid derivative (Richardson et al., 2017) |
| 16aa Compound identified from Brassica juncea . | Methylthiobenzoic acid |  |
(4‐Methylthiobenzoic Acid) Anti nephrotoxicity, antioxidant (Husain et al., 1996) |
| 17aa Compound identified from Brassica juncea . | Polidocanol |  |
Sclerosing agent (Goldman & Guex, 2017) |
| 18bb Compound identified from Solanumtuberosum . | 3‐Methylbutyraldehyde oxime |  |
Plant derivative (Sørensen et al., 2018) |
| 19bb Compound identified from Solanumtuberosum . | 6‐Hydroxy pseudo oxynicotine |  |
Nicotine metabolite (Ganas et al., 2008) |
| 20bb Compound identified from Solanumtuberosum . | 8‐Isoquinoline methanamine | 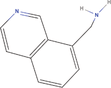 |
Isoquinoline derivative (You et al., 2019) |
| 21bb Compound identified from Solanumtuberosum . | Desethyletomidate | 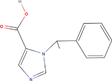 |
Etomidate derivative and 11β‐Hydroxylase inhibitor (Pejo et al., 2016) |
| 22bb Compound identified from Solanumtuberosum . | DMPO | 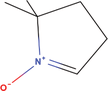 |
Spin trapping reagent for free radical detection (Kalyanaraman et al., 1994) |
| 23bb Compound identified from Solanumtuberosum . | Gallic acid | 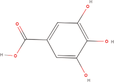 |
Natural phenolic compound (Kahkeshani et al., 2019) |
| 24bb Compound identified from Solanumtuberosum . | N‐(2‐Methylpropyl) acetamide |  |
Acetamide derivative found alcoholic beverages (Yannai, 2003) |
| 25bb Compound identified from Solanumtuberosum . | N ,N‐dimethyl‐Safingol | 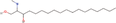 |
Sphingolipid derivative for anti‐inflammatory action (Vasconcelos et al., 2017) |
| 26bb Compound identified from Solanumtuberosum . | N‐methyl hexanamide |  |
Hexamide derivative (Sha et al., 1995) |
| 27bb Compound identified from Solanumtuberosum . | Phytosphingosine | 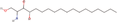 |
Antimicrobial and anti‐inflammatory natural compound (Pavicic et al., 2007) |
| 28bb Compound identified from Solanumtuberosum . | Pinidine |  |
Piperidine alkaloid (Virjamo et al., 2010) |
| 29bb Compound identified from Solanumtuberosum . | Sphinganine | 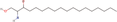 |
Sphingolipid derivative (Christie & Han, 2012) |
| 30bb Compound identified from Solanumtuberosum . | Uzarigenin | 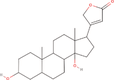 |
Uzarigenin is a component of Uzara, which has been used for a long time in traditional medicine to treat diarrheal disorders (Schulzke et al., 2011) |
| 31cc Reference molecule. | Hydroxychloroquine | 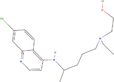 |
Treatment of malaria and rheumatoid arthritis (Ben‐Zvi et al., 2012) |
- a Compound identified from Brassica juncea .
- b Compound identified from Solanumtuberosum .
- c Reference molecule.
2.2 Molecular docking
Protein targets were downloaded from database, Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do). TMPRSS2 (PDB: 1Z8G) (Sakai et al., 2014), SARS‐CoV‐S in complex with ACE2 (PDB: 2AJF) (Li et al., 2005; Micholas & Jeremy, 2020), PLpro (PDB: 3E9S) (Wu et al., 2020), SARS‐CoV‐2 3CLpro (PDB: 6LU7) (Wu et al., 2020), SARS‐CoVRdRp (PDB: 6NUR) (Elfiky, 2020), SARS‐CoV‐2 spike glycoprotein structure (SARS‐CoV‐2‐S) (PDB: 6VSB) (Grifoni et al., 2020), binding complex of human ACE2 and RBD (PDB: 6VW1) (Qiu et al., 2020), SARS‐CoV‐2‐S (closed) (PDB: 6VXX) (Walls et al., 2020), SARS‐CoV‐2‐S with one SB(open) (PDB: 6VYB) (Walls et al., 2020) and Human angiotensin‐converting enzyme 2 (ACE2) (PDB: 1R42) (Jia et al., 2009) are selected for the present study as target proteins (Figure 1). All water molecules were removed, and in the final stage, hydrogen atoms were added to the receptor molecule. For the ligand preparations, SMILES was obtained from PubChem and was further translated to mol file using “Open babel” Translator A Molecular Mechanics (MM) method UFF was used for refining initial geometries, using the “Clean Geometry” option in the ArgusLab Software. The active site was defined from the coordinates of the ligand in the original PDB files Protein for the receptor protein. Residues that lie within 5 Å unit area of ligand that interact with it through their side chain were identified and were considered as Active site residues. The docking between receptor and ligand was performed using the “Dock a ligand” command. A spacing of 0.4 Å between the grid points was used. Binding site box size was set to (15 × 15 × 15 Å) so as to encompass the entire active site. Molecular docking was implemented on 1Z8G, 2AJF, 3E9S, 6LU7, 6NUR, 6VSB, 6VW1, 6VXX, 6VYB and 1R42 receptor against ligands using Argus Lab 4.0.1 (Mark A. Thompson, Planaria Software LLC, Seattle, WA, http://www.arguslab.com) to find the reasonable binding geometries and to explore the protein–ligand interactions. Docking of the protein–ligand complex was mainly targeted only on the predicted active site. Docking simulations were performed by selecting “Argus Dock” as the docking engine and their relative stabilities were evaluated using molecular dynamics, and their binding affinities, using free energy simulation, pose and time. Single‐trajectory method was used for the binding energy calculation. The selected residues of the receptor were defined to be a part of the binding site and ligand–protein interaction visualized by Biovia Discovery studio.

3D structures of selected proteins as a receptor for the virtual screening (a) 1Z8G, (b) 2AJF, (c) 3E9S, (d) 6LU7, (e) 6NUR, (f) 6VSB, (g) 6VW1, (h) 6VXX, (i) 6VYB and (j) 1R42 [Colour figure can be viewed at wileyonlinelibrary.com]
3 RESULTS
The field of molecular docking has emerged during the last three decades and now is becoming an integral aspect in drug discovery and development area. Molecular docking is utilized for the prediction of protein–ligand complexes, which is composed of two components: a search algorithm, an algorithm that creates possible protein–ligand complex geometries, and thus performs the process of “pose generation,” and a scoring function that predicts the binding affinity of the ligand to the protein based on the complex geometry. Binding energies are the most widely used mode of measuring binding affinity of a ligand. In the present study, binding energy of the best pose was recorded as docking results of the selected compounds, carried out by Argus lab in Table 2.
| Sr. no. | Name | Docking score (ΔG kcal/Mol) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1Z8G | 2AJF | 3E9S | 6LU7 | 6NUR | 6VSB | 6VW1 | 6VXX | 6VYB | 1R42 | ||
| 1. | Curcumenol | −7.62 | −5.88 | −7.09 | −6.33 | −6.86 | −6.43 | −6.31 | −6.62 | −6.75 | — |
| 2. | N‐Desmethylselegiline | −7.13 | −5.35 | −8.36 | −5.99 | −6.53 | −6.28 | −4.5 | −6.35 | −6.22 | −9.77 |
| 3. | Phentermine | −7.19 | −5.77 | −7.93 | −6.28 | −6.92 | −6.25 | −4.85 | −6.44 | −6.43 | −9.8 |
| 4. | Monodesmethylpheniramine | −7.18 | −5.15 | −10.55 | −6.16 | −6.32 | −6.53 | −5.39 | −7.31 | −5.87 | — |
| 5. | N‐(3E‐hexadecenoyl)‐deoxysphing‐4‐enine‐1‐sulfonate | −10.28 | — | −10.88 | −5.31 | −6.49 | — | −2.55 | — | −5.19 | — |
| 6. | (5alpha,8beta,9beta)‐5,9‐Epoxy‐3,6‐megastigmadien‐8‐ol | −7.5 | −5.85 | −6.99 | −6.27 | −6.71 | −6.62 | −6.24 | −6.54 | −6.02 | −7.19 |
| 7. | (R)‐2,3‐Dihydroxypropane‐1‐sulfonate | −6.23 | −6.03 | −5.67 | −6.26 | −6.42 | −6.34 | −6.57 | −5.57 | −5.92 | −9.75 |
| 8. | 2‐(3‐Phenylpropyl) tetrahydrofuran | −6.63 | −4.84 | −7.48 | −6.46 | −6.63 | −5.82 | −6.64 | −6.65 | −6.89 | — |
| 9. | 3‐Nonyl‐1H‐pyrazole | −6.88 | −5.97 | −6.77 | −5.71 | −6.19 | −5.65 | −4.87 | −6.36 | −5.55 | −9.91 |
| 10. | Mephentermine | −7.08 | −5.31 | −7.55 | −5.83 | −6.55 | −5.83 | −4.63 | −6.32 | −6.07 | — |
| 11. | N‐(2,4‐Eicosadienoyl) piperidine | −7.44 | −5.05 | −8.1 | −5.59 | −6.42 | −5.7 | −3.27 | −6.73 | −6.19 | — |
| 12. | Benzocaine | −6.05 | −5.3 | −6.46 | −5.87 | −6.33 | −5.62 | −5.5 | −5.99 | −5.39 | −8.55 |
| 13. | CAY10638 (5‐(4‐(2‐[Thiophen‐2‐yl]ethoxy)benzylidene)thiazolidine‐2,4‐dione) | −6.2 | −4.97 | −7.56 | −5.08 | −6.85 | −5.93 | −4.55 | −6.68 | −6.63 | — |
| 14. | Dimethylallyl diphosphate (DMAPP) | −6.69 | −5.86 | −6.77 | −6.47 | −7.01 | −5.63 | −4.22 | −6.01 | −6.52 | — |
| 15. | Î ± ‐9(10)‐EpODE | −7.68 | −5.19 | −7.64 | −5.59 | −6.46 | −5.73 | −6.06 | −6.22 | −5.41 | — |
| 16. | Methylthiobenzoic acid | −6.29 | −4.95 | −6.99 | −6 | −6.16 | −6.22 | −4.78 | −6.53 | −5.96 | −8.54 |
| 17. | Polidocanol | — | −2.95 | — | −4.24 | — | −3.54 | −3.13 | −4.04 | — | −5.75 |
| 18. | 3‐Methylbutyraldehyde oxime | −6.02 | −5.18 | −6.19 | −5.36 | −5.96 | −6.07 | −5.58 | −5.64 | −5.58 | −8.2 |
| 19. | 6‐Hydroxy pseudo oxynicotine | −6.08 | −5.41 | −6.56 | −5.82 | −6.41 | −6.6 | −5.47 | −5.73 | −5.27 | — |
| 20. | 8‐Isoquinoline methanamine | −6.83 | −6.01 | −7.11 | −6.48 | −6.84 | −6.81 | −5.5 | −6.53 | −6.09 | — |
| 21. | Desethyletomidate | −6.44 | −5.29 | −8.19 | −5.99 | −6.68 | −5.85 | −4.68 | −6.31 | −5.89 | — |
| 22. | DMPO | −5.77 | −5.16 | −5.67 | −5.38 | −5.84 | −5.69 | −4.26 | −5.42 | −7.09 | −7.16 |
| 23. | Gallic acid | −5.22 | −5.03 | −5.63 | −5.3 | −5.67 | −5.37 | −5.67 | −5.4 | −6.09 | — |
| 24. | N‐(2‐Methylpropyl) acetamide | −5.81 | −5.09 | −6.15 | −5.42 | −5.74 | −5.59 | −6.28 | −5.75 | −6.33 | −7.06 |
| 25. | N ,N‐dimethyl‐Safingol | −6.17 | −5.64 | −7.09 | −4.77 | −5.9 | −5.32 | −4.58 | −5.92 | −6.07 | — |
| 26. | N‐methyl hexanamide | −5.81 | −4.96 | −6.31 | −5.54 | −5.58 | −5.38 | −4.71 | −5.8 | −5.1 | −7.63 |
| 27. | Phytosphingosine | −6.95 | −5.39 | −6.86 | −6.32 | −5.56 | −5.41 | −3.64 | −6.57 | −4.99 | — |
| 28. | Pinidine | −6.73 | −5.43 | −6.18 | −5.71 | −6.08 | −6.35 | −5.41 | −5.99 | −5.8 | −8.38 |
| 29. | Sphinganine | −9.3 | −4.62 | −6.82 | −5.61 | −6.1 | −5.55 | −3.78 | −6.29 | −5.11 | — |
| 30. | Uzarigenin | −7.87 | −5.95 | — | −5.37 | — | −7.39 | −5 | −4.25 | −5.78 | — |
| 31. | Hydroxychloroquine | −6.9 | −5.59 | −6.54 | −5.78 | −5.61 | −5.94 | −5.54 | −5.35 | −5.12 | — |
- Note: Desh (—) symbol indicates no acceptable ligand poses were found.
From the virtual screening, with the proteins of human and SARS‐coronavirus, involved in the mechanism of infection, three most potent compounds were found, namely (a) curcumenol, (b) N‐desmethylselegiline and (c) Phentermine exhibited promising activity based on the docking score (Table 2). Moreover, Monodesmethylpheniramine and N‐(3E‐hexadecenoyl)‐deoxysphing‐4‐enine‐1‐sulfonate also possess remarkable activity with high specificity and selectivity towards the selective proteins (Table 2). Wherever the structure of ligands not properly fit in targeted receptors, software shows the message as “No acceptable ligand poses were found.” Molecular docking analysis of curcumenol with 3E9S showed interactions with Val126, Phe128, Gln134, Glu135, Arg141 and CL319 with van der walls, Lys127 with Alkyl and Tyr138 with Pi‐Alkyl; 6NUR with His295, Leu302 and Cys306 with Alkyl, Arg305 and His309 with Pi‐Alkyl and Gln292, Tyr294, Asn300, Cys301, Asp303, Cys310 and ZN1001 with van der Walls; 6VYB with Asn61 with Covalent bond and Tyr28, Thr29, Asn30 and Asn61 with van der Walls; and 1Z8G with His203 with covalent bond, Gly388 and Csy381 with C‐H bond, Tyr301, Asp347, Gln350, Gly351, Ser353, Ser376, Gly378, Thr379 and Gly380 with conventional H bond and Trp327 with Pi‐Alkyl at various bond lengths as shown in Figure 2a. The docking study of N‐desmethylselegiline with 1Z8G with Ser353, Gly351, Ser376, Gln350, Asp347, Gly380, Tyr301, Thr379 and Gly378 with conventional H bond, Gln331, Asn254, Tyr243, Asp352, Val375, Cys349, Val389, Ala348 and Ala382 with van der Walls, Gly388 and Cys381 with CH bond, His203 with Pi‐Sigma and Trp377 with Pi‐Alkyl; 6NUR with His309, Thr293, Asp303, His295 and Cys301 with van der Walls, Cys306 with Alkyl, Arg305 and Leu470 with Pi‐Alkyl and Leu302 with Pi‐Sigma; 6VYB with Tyr28, Thr29 and Asn30 with van der Walls and Asn61 with Covalent bond and 1R42 with Tyr613, Unk909 and Unk911 with van der Walls and Asp615, Unk907, Unk908 and Unk910 with conventional H bond; and 3E9S with Gln134 with C‐H bond, Leu121, Glu135, Arg141 and Lys127 with van der Walls, Phe128 with Pi‐Pi stalked and Tyr138 with Pi‐Alkyl (Figure 2b). The docking analysis of phentermine with 1Z8G with Ser353, Gly351, Ser376, Gln350, Asp347, Gly380, Tyr301, Thr379 and Gly378 with conventional H bond, Asn254, Gln331, Ala382, Tyr243, Asp352, Val375, Cyc349, Val389, Ala348 and Cys381 with van der Walls, Trp377 with Pi‐Alkyl and His203 with Pi‐Sigma; 3E9S with Tyr137, Gln134, Glu135, Arg139 and Ala136 with van der Walls; 6NUR withAsn300, His295, Thr293, His309, Gln292, Arg305, Leu470, Asp303 and Cys301 with van der Walls, Cys306 with Pi‐Alkyl and Leu302 with Pi‐Sigma; 1R42 with Tyr613, Unk909 and Unk911 with van der Walls and Unk907, Unk908, Asp615 and Unk910 with Conventional H bond; and 6VYB with Asn30, Thr29 and Tyr28 with van der Walls and Asn61 with covalent bond (Figure 2c).

Low‐energy binding confirmation shown as a surface model with detailed view bound to (a) curcumenol with 1Z8G, (b) N‐desmethylselegiline with 3E9S and (c) Phentermine with 6VYB by molecular docking [Colour figure can be viewed at wileyonlinelibrary.com]
In addition, other compounds brought promising results of binding activity against the studied proteins, such as (5alpha,8beta,9beta)‐5,9‐Epoxy‐3,6‐megastigmadien‐8‐ol, (R)‐2,3‐Dihydroxypropane‐1‐sulfonate, 2‐(3‐Phenylpropyl) tetrahydrofuran, 3‐Nonyl‐1H‐pyrazole, 6‐Hydroxy pseudo oxynicotine, N‐(2,4‐Eicosadienoyl) piperidine, pinidine, sphinganine and uzarigenin showed remarkable binding efficiency with different proteins. Results of the study strikingly endorse the evaluation of sphingolipid derivatives as a treatment of COVID‐19, as we have found various fatty acid and sphingolipid derivatives with low energy binding score with studied proteins. HCQ exhibited less specificity in binding with proteins selected in the present study, in comparison to the three promising molecules from docking score, that is, (a) curcumenol, (b) N‐desmethylseleginine and (c) phentermine.
4 DISCUSSION
COVID‐19 therapies follow different approaches, that is, blocking an enzyme function requires virus multiplication, blocking attachment site of viral proteins on human cell receptor, enhancing host immunity and blocking the host cell receptor/proteins to block the virus attachment. Protein–protein interaction, in terms of receptor–ligand recognition, is the general mechanism of virus infection in host cell. SARS‐CoV‐S protein recognized the ACE2 receptor of host cell and facilitates the ingestion of viral components (Li et al., 2003). Blocking the interaction between host ACE2 and SARS‐CoV‐S can be utilized as a strategy to prevent the COVID‐19 infection in host cell. Human ACE2 recognition as a receptor by SARS‐CoV‐2‐S is due to its structural similarity and sequence conservation with SARS‐CoV‐S glycoproteins (Walls et al., 2020). Moreover, structural studies of ACE2 in complex with SARS‐CoV‐2‐S, with human and other species may contribute to understanding interspecies transmission and receptor usage of COVID‐19 (Qiu et al., 2020). In SARS‐CoV infection, ACE2 is released from human airway epithelial cells, an important initial site of infection, where it retains terminal carboxypeptidase activity as well as its ability to bind the virus, in addition, ACE2 must be cell attached to function as a SARS‐CoV receptor (Jia et al., 2009). Previously, it was demonstrated that TMPRSS2 mediated ACE2 cleavage increases SARS‐CoV‐S‐mediated entry in host cell (Heurich et al., 2014; Shulla et al., 2011). Therefore, targeting the TMPRSS2 is also important as a preventive strategy against COVID‐19 infection.
Curcumenol was studied earlier for its interaction with human serum albumin through the docking study (Hamdi et al., 2015), exhibiting molecular interaction and ligand selectivity with human protein. In the present result, curcumenol showed binding with TMPRSS2, ACE2 complexed with SARS‐CoV, SARS‐CoV‐S, PLpro, 3CLpro and RdRP of SARS‐CoV, as well as SARS‐CoV‐2‐S (closed and open), looking into the docking score of these proteins with curcumenol gives insight of selectivity towards bound human proteins. In contrast, ACE2 remained without any interaction with curcumenol, which indicates the selectivity of curcumenol towards bound human receptors with SARS‐CoV. Results of the docking study need to be further confirmed through protein–protein interaction study, in vitro and in vivo , to treat infected patients. This confirmed the selectivity of curcumenol towards human receptor complexes with COVID‐19 proteins. However, curcumenol showed remarkable docking score with open SARS‐CoV‐2‐S compared to the closed state, which further supports the preventive effect of the curcumenol to healthy uninfected individuals to suppress the binding capacity of SARS‐CoV‐2 in human host. This finding directs the repurposing of curcumenol as a drug after in vivo and in vitro confirmation.
We have found N‐desmethylselegiline is a metabolite of selegiline (Monoamine oxidase‐B inhibitor) used for the treatment of Parkinson’s disease (Heinonen et al., 1994) and protects neurons through stimulating expression of the neurotrophic factors (Mizuta et al., 2000). N‐Desmethylselegiline interacted with all the studied proteins with remarkable docking score, mainly TMPRSS2, SARS‐CoV‐2‐S, PLpro, RdRp and ACE2 are involved in the mechanism of infection of SARS‐CoV‐2 in human (Hoffmann et al., 2020). Spike protein cleaved into S1 and S2 by the host cell, TMPRSS2, and other proteases, and S1 binds with host cell surface receptors, whereas S2 mediates virus–cell and cell–cell membrane fusion (Wu et al., 2020). Spike structural integrity and cleavage activation play a key role in virus invasion and virulence (Wu et al., 2020). TMPRSS2 activates SARS‐CoV infection through spike protein of virus in host cell, so the inhibition of TMPRSS2 can prevent the coronaviruses entry into host cells (Glowacka et al., 2011). Recently, to target SARS‐CoV‐2, many anti‐viral and anti‐bacterial drugs are virtually screened to inhibit TMPRSS2 (Wu et al., 2020). Present results complement the protease specific action of the drug, binding to PLpro showed the preventive effect/inhibitory effect to virus infection, encouraged the use as an ACE2 inhibitor as reflected from the docking score.
Phentermine has been approved by the Food and Drug Administration (FDA) as appetite suppressants for the treatment of obesity (Connolly et al., 1997). Phentermine effects involve dopamine neurons, which increase extracellular dopamine and involve in appetite control (Baumann et al., 2000). Phentermine showed interaction with minimum energy level with TMPRSS2, PLpro. RdRp and ACE2. Phentermine was found to bind with all the studied protein structures of human and SARS‐CoV, as well as its complexes, compliant with its potency and selectivity to block the coronavirus infection in human.
The docking score of Monodesmethylpheniramine with TMPRSS2 and PLpro, as well as the closed structure of SARS‐CoV‐2‐S, revealed its role in blocking the initial infection state of SARS‐CoV‐2. However, Monodesmethylpheniramine did not show any interaction with ACE2 related carboxypeptidase; it revealed the role in inhibition of infection over the unbound protein complex. The present findings of the docking score of Monodesmethylpheniramine interaction with PLpro to block the SARS replication endorse in vivo and in vitro examination of Monodesmethylpheniramine as a COVID‐19 treatment. Moreover, chlorpheniramine (A halogenated form of pheniramine) is found to be deposited in the lung (Kamm et al., 1969), which directs the present research for nasal spray based drug delivery of pheniramine family drug in patients with SARS‐CoV‐2 infection.
In the present study, N‐(3E‐hexadecenoyl)‐deoxysphing‐4‐enine‐1‐sulfonate, a sphingolipid, showed remarkable binding with TMPRSS2 and PLpro, indicating the role of sphingolipids in binding with human and SARS‐CoV‐2 proteins. Sphingolipid showed activity to reduce inflammation and apoptosis of lung cell in different pulmonary complications (Sharma & Prakash, 2017), and sphingolipid can be a target for the treatment of pulmonary infection, as reflected in our results. Likewise, 3‐Nonyl‐1H‐pyrazole, an important natural pyrazole derivative, is a nitrogen‐containing five‐membered heterocyclic compound and exhibits effectiveness in the field of drug discovery and antimicrobial activity (Kumar et al., 2013; Vijesh et al., 2013). Molecular docking study of the pyrazole‐based drug with DNA gyrase (Shubhangi Kumar et al., 2019) complements our results of molecular docking of human and SARS‐CoV‐2 proteins.
A repurposing of drug for SARS‐CoV‐2 treatment is in practice to control the infection and replication of the virus. Antimalarial hydroxychloroquine (HCQ) has been found effective against SARS–CoV‐2 in vitro , as well as in controlled or uncontrolled clinical studies (Gautret et al., 2020; Liu et al., 2020; Yao et al., 2020). Moreover, HCQ has been recommended in India to treat suspected and confirmed cases of COVID‐19 (COVID‐19, 2020). In addition, HCQ use has to be balanced against the risk of cardiac arrhythmia (Chen et al., 2006), whereas curcumenol and N‐desmethylselegiline have not been reported of side effects on heart function. In contrast, Phentermine is associated with valvular heart disease—upon chronic exposure (Connolly et al., 1997). Apart from the best three molecules found in the present study, other molecules from natural sources like (5alpha,8beta,9beta)‐5,9‐Epoxy‐3,6‐megastigmadien‐8‐ol, (R)‐2,3‐Dihydroxypropane‐1‐sulfonate, 2‐(3‐Phenylpropyl) tetrahydrofuran and 3‐Nonyl‐1H‐pyrazole showed remarkable activity against all the proteins, including ACE2, SARS‐Cov‐2‐S and TMPRSS2. Here, it is reported that the molecules from plant source show chances of less adverse effects compared to other xenobiotics. Both plants as a source of the compounds are regular nutraceutical and can be utilized further in pharmaceutical formulations in combination with other potential derivatives.
The entire experiment uncovered the probable efficacy of previously identified drug molecules as well as compounds from plant source to find the quick, effective, broadly specific and with minimum adverse/side effects to combat COVID‐19 pandemic. Furthermore, comparative evaluation of the studied molecules with HCQ endorsed the in vitro experiments against SARS‐CoV‐2 to establish the potent medication. Repurposing of our studied molecules over the HCQ needs to be examined in vitro and in vivo . In nutshell, results provide a basic novel protein–ligand interaction framework to identify promising molecule for ongoing drug discovery and immediate therapeutics against COVID‐19.
In summary, the results of the studied compounds showed utility at various levels of COVID‐19 infection. COVID‐19 infection mechanism involves binding with ACE2 and TMPRSS2 receptor on the cell surface of host, followed by entry and release of viral RNA and protein machinery in host cell. To prevent the host‐specific entry through binding with the receptor on cell surface, the present work and identified compounds proved their activity against various mode of action, that is, (a) binding inhibition with cell surface receptor, (b) masking the viral or host protein to prevent the attachment and (c) making the host–virus binding complex inactive. We recommend to develop a medication of spray formulation and nasal route of administration to treat the COVID‐19 patients with minimum side effects and improvement in recovery time. Since the compounds are from plant source, the study further supports Ayurveda based preventive formulations against the SARS‐CoV‐2, utilizing the plant materials in different extracts. However, further i n vitro and in vivo , as well as toxicological studies, are required to validate and develop the application of promising molecules for the prevention, cure and treatment of COVID‐19.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
REFERENCES